Please verify you are a human
Access to this page has been denied because we believe you are using automation tools to browse the website.
This may happen as a result of the following:
- Javascript is disabled or blocked by an extension (ad blockers for example)
- Your browser does not support cookies
Please make sure that Javascript and cookies are enabled on your browser and that you are not blocking them from loading.
Reference ID: #ee9ff66c-ae59-11ee-b45a-c499c99f108d
Powered by PerimeterX , Inc.
5 quants / Лабораторный практикум Квантово-химическое моделирование соединений в пакете HyperChem Учебно-методическое пособие
Высшего профессионального образования «Кемеровский государственный университет» Кафедра экспериментальной физики Лабораторный практикум Квантово-химическое моделирование соединений в пакете HyperChem Учебно-методическое пособие
Кемерово 2013
Составитель: к.ф.-м.н., доцент А.Л. Юдин Лабораторный практикум «Квантово-химическое моделирование соединений в пакете HyperChem»: учеб.-метод. пособие / ФГБОУ ВПО «Кемеровский государственный университет»; сост. А.Л. Юдин. – Кемерово, 2013. – 175 с. Цель пособия – выработать у студентов навыки решения практических задач квантовой химии с использованием современного программного обеспечения на примере комплекса HyperChem. Приведено описание вычислительных возможностей квантовохимического пакета HyperChem. Рассмотрены правила и примеры, позволяющие на практике проводить молекулярно-механические, неэмпирические и полуэмпирические расчеты геометрии, электронных состояний и различных свойств молекул. Даны методические указания и рекомендации по выполнению лабораторных работ в HyperChem. Приведено обсуждение полученных результатов и даны примеры анализа конкретных молекул. Пособие подготовлено на кафедре экспериментальной физики и предназначено для студентов физического факультета, обучающихся по направлению 011200.62 «Физика» и выполняющих лабораторный практикум по курсу «Математическое моделирование структуры и свойств химических соединений». Пособие может использоваться и для других специальных курсов, связанных с изучением и применением расчетных методов квантовой химии, а также для работы над курсовыми и дипломными проектами по данному направлению.
| РЕКОМЕНДОВАНО | РАССМОТРЕНО |
| методической комиссией | на заседании кафедры |
| физического факультета | экспериментальной физики |
| Протокол № 3 | Протокол № 4 |
| от «12» ноября 2012 г. | от «15» ноября 2012 г. |
| Председатель методической | Заведующий кафедрой |
| комиссии | _________________________ |
| _______________________ | |
| Н. И. Гордиенок | С. Д. Шандаков |
© Кемеровский государственный университет © Юдин А.Л. 2
I. ПАКЕТ КВАНТОВО-ХИМИЧЕСКИХ РАСЧЕТОВ HYPERCHEM 1. Введение В настоящее время компьютерное моделирование методами квантовой химии и молекулярной динамики электронной и атомной структур молекулярных и кластерных систем переходных наноразмеров различной сложности получило широкое распространение. Квантово-химические методы с большой точностью, не прибегая к дорогостоящему эксперименту, позволяют не только рассчитать свойства отдельных молекулярных систем, но и выявлять общие закономерности, присущие классам соединений, обосновывать существующие закономерности, проводить корреляции [1-14]. Как известно, в основе квантово-химических расчетов лежит решение уравнения Шредингера. Однако даже для многоэлектронных атомов и тем более для многоатомных систем решение уравнения точным численным методом неприемлемо из-за очень большого расчетного времени. Точное решение невозможно даже для молекулы водорода. Последовательное применение приближенного метода решения уравнений Хартри-Фока-Рутаана — метода самосогласованного поля (ССП) — для молекул, состоящих всего из нескольких атомов, может натолкнуться на непреодолимые вычислительные проблемы. По этой причине в квантовой химии значение приобретают полуэмпирические (приближенные) методы решения этого уравнения. Поиск путей решения уравнения Шредингера для молекул в рамках схемы ССП может идти по нескольким направлениям. В зависимости от степени приближения все квантово-химические методы можно разделить на неэмпирические (первопринципный ab initio (от лат. « от начала ») и функционала плотности), полуэмпириче- ские , эмпирические (группа методов молекулярной механики ) и методы молекулярной динамики . На сегодняшний день в мире существует много вычислительных комплексов и программных продуктов, в которых реализованы методы квантовой химии. Наиболее полный список программ с описанием возможностей представлен в работе [15]. Широко используются такие пакеты, как Gaussian ( www.gaussian.com ), Gamess ( www.msg.ameslab.gov/GAMESS ), ChemCraft, как средство визуа-
лизации программ расчетных файлов Gaussian и Gamess ( www.chemcraftprog.com ), MOPAC ( openmopac.net ), Molden и мно- гие другие. К таковым относится и пакет программ HyperChem , возможности которого будут рассмотрены в данном пособии. Бесплатную демонстрационную 10-дневную, но полнофункциональную, версию программы можно получить на сайте компании Hypercube ( www.hyper.com ). Программа снабжается документацией (на английском языке) в файлах: CDK.pdf, GetStart.pdf, Referenc.pdf (основаны на версии 5.0), hyperchem_getting_started.pdf (для версии 7.5), hyper8.pdf (описание текущей версии 8.0). HyperChem – комплексный программный продукт, предназначенный для задач квантово-механического моделирования атомных и молекулярных структур (рис. 1.1). Он включает в себя программы, реализующие методы молекулярной механики, квантовой химии и молекулярной динамики. Силовые поля молекулярной механики, которые могут использоваться в HyperChem – это ММ+, Amber, OPLS и BIO+ (на базе CHARMM). Реализованы полуэмпирические методы: расширенный метод Хюккеля, CNDO, INDO, MINDO/3, MNDO, AM1, PM3, ZINDO/1, ZINDO/S и другие. Представлены не- эмпирические методы расчета: ab initio (Хартри-Фока) и метод функционала плотности. Все результаты квантово-химических расчетов, представленные в данном пособии, могут быть получены с использованием различных версий программы HyperChem старше 6.0. Приведенные ниже практические инструкции по работе с программой основаны на имеющейся у автора коммерческой версии HyperChem 8.07. Пакет программ HyperChem позволяет проводить неэмпирические и полуэмпирические расчеты геометрических, электронных, спектральных и магнитных характеристик молекул, межмолекулярных комплексов и наноструктур, а также вычислять энергию переходных состояний комплексов, расчеты электронных и колебательных спектров, характеристики систем в гидратной (сольватной) оболочках, производить простейшие расчеты характеристик кристаллов. Данный пакет предназначен в первую очередь для решения вычислительных задач компьютерной химии, нанофизики. Во многих случаях имеется возможность сопоставления результатов расчетов с экспериментальными данными.
Рис. 1.1 . Общий вид окна программы HyperChem (представлено изображение нанотрубки) Информационное содержание квантово-химических расчётов выше, чем информативность эксперимента. Они позволяют одновременно рассчитать геометрию молекул, дипольные моменты, энтальпии, потенциалы ионизации, распределение зарядов, порядки связей, спиновые плотности и т.п. Причём эти данные можно получить и для несуществующих в действительности молекул. Можно прогнозировать некоторые характеристики для гипотетических молекул, не прибегая к эксперименту. Последняя версия HyperChem 8.0 содержит, как и предыдущие, графический редактор, большую базу данных по строению молекул, наносистем, нуклеотидов, полимеров, элементарных ячеек кристаллов и способна считать всеми современными методами квантовой химии. База данных позволяет легко построить структуры белков, полимеров, фрагменты ДНК, нанокластеры металлов, металлоорганических соединений, углеродные наноструктуры. Достаточно большой набор различных методов молекулярной механики, полуэмпирических методов и всевозможные типы базисов, используемые в ab initio расчетах, включая расщепленные и поляризованные, обеспечивают широкий спектр вычислений в 5
HyperChem . К сожалению, ab initio расчеты даже в базисе 3-21G требуют значительных ресурсов оперативной, общей памяти ЭВМ, большой мощности процессора. В настоящее время этот метод редко применяется для молекул или молекулярных фрагментов, содержащих более 10 атомов. Самым несомненным преимуществом программы HyperChem является возможность наглядного изображения графической структуры молекулы (рис. 1.1) и изменение геометрических параметров при оптимизации системы в интерактивном режиме. Также программа отличается возможностями визуализации полученных в результате расчетов: молекулярных орбиталей, относительной интенсивности электронных переходов, потенциалов в двумерном изображении и анимация колебательных мод. При изложении материала предполагалось, что читатель знаком с основами химического строения веществ, университетским курсом атомной физики, а также с базовыми основами квантовой механики и квантовой химии. И хотя пособие ориентировано на студентов физиков, не специализирующихся на квантовой химии, квантовохимические программы нельзя использовать как некий «черный ящик», не понимая идеи и методы современной квантовой химии. Необходимо также отметить, что для успешного освоения программы пользователю потребуются базовые навыки работы в операционной системе Windows. 2. Использование графического режима в пакете HyperСhem 2.1. Рабочее окно HyperChem Главное окно программы HyperChem представлено на рис. 1.2. Окно программы, являющееся стандартным окном Windowsприложения, содержит строку названия, строку меню, панель инструментов, строку состояния и основное рабочее поле визуализации и построения, где находится текущая молекулярная система. Строка названия вверху экрана показывает имя файла, с которым Вы работаете. Если Вы работаете во вновь созданном файле, появляется имя untitled . А также здесь расположены кнопки управления окном, которые содержат команды, позволяющие менять размеры, перемещать, расширять и закрывать окно HyperChem .
Строка меню содержит имена ниспадающих списков команд основного HyperChem меню: File (Файл), Edit (Редактирование), Build (Построение объектов), Select ( Выделение), Display (Экран), Databases (Базы данных), Setup (Установки), Compute (Расчет), Annotations ( Подписи), Script (Сценарий), Cance l (Отмена), Help (По- мощь). Рис. 1.2. Элементы рабочего окна HyperChem Панель инструментов Левая сторона строки инструментов содержит восемь кнопок инструментальных средств, которые Вы используете, чтобы построить, выбрать, отобразить, и переместить атом или молекулу  . Правая сторона содержит набор стандартных кнопок
. Правая сторона содержит набор стандартных кнопок  , выполняющих типичные действия с файлами и объектами в Windowsприложениях. В рабочем поле отображаются все загружаемые или создаваемые исследователем структуры. По умолчанию рабочее поле имеет черный фон. При желании цвет фона можно изменить, например, на
, выполняющих типичные действия с файлами и объектами в Windowsприложениях. В рабочем поле отображаются все загружаемые или создаваемые исследователем структуры. По умолчанию рабочее поле имеет черный фон. При желании цвет фона можно изменить, например, на
более привычный белый, в пункте Preferences ( Настройки ) главного меню File ( Файл ). Строка состояния в нижней части экрана показывает в зависимости от режима работы программы текущую информацию, такую как количество атомов в молекуле, состояние, результаты вычисления или ход вычислительного процесса, энергию или величину градиента, длины выбранных связей или углов, подсказки и т.п. Когда Вы выбираете пункт меню, краткое описание пункта появляется в строке состояния. В правом углу строки состояния высвечивается сокращенное название выбранного вычислительного алгоритма (метода расчета), который в данный момент используется программой. Принцип работы программы таков, что для конкретной молекулярной модели, загруженной в окне визуализации, можно решать однотипные задачи, например, минимизировать энергию молекулы, используя различные методы. Выбор метода производится в пункте Setup основного меню. В частности, словосочетание Ab Initio в правом нижнем углу экрана на рис. 1.1 означает, что рабочим расчетным методом в данный момент является данный неэмпирический квантовохимический метод. На рисунке 1.2 выбран метод ММ+ . 2.2. Использование мыши и инструментов Работа в HyperChem по большей части происходит при помощи мыши. Она используется следующим образом: 1. L-Нажатие — щелчок левой кнопкой мыши. 2. R-Нажатие — щелчок правой кнопкой мыши. Обычно, R- нажатие имеет противоположный эффект L-нажатию. 3. Двойное нажатие — быстрый двойной щелчок левой кнопкой мыши. 4. L-протяжка или R-протяжка — нажатие на левую или правую кнопку мыши, и перемещение, не отпуская кнопки (скольжение), курсора в новую позицию в рабочей области. 5. LR-протяжка — нажатие и удержание левой кнопки мыши, затем правой кнопки и перемещение курсора в новую позицию в рабочей области. 6. RL-протяжка — так же, как LR-протяжка, но вначале происходит нажатие правой кнопки мыши. Курсор мыши способен изменять форму. Чтобы это увидеть:
1. Нажмите левую клавишу на инструментальном изображении  ( Draw — рисовать ). 2. Переместите курсор в рабочую область. Курсор приобретает соответствующую форму: . 3. Укажите на инструментальное изображение
( Draw — рисовать ). 2. Переместите курсор в рабочую область. Курсор приобретает соответствующую форму: . 3. Укажите на инструментальное изображение  ( Select — выбор ) и нажмите левую клавишу. 4. Переместите курсор в рабочую область. Он приобретает соответствующую форму: . Таким же образом изменяется форма курсора при выборе остальных шести кнопок строки инструментов.
( Select — выбор ) и нажмите левую клавишу. 4. Переместите курсор в рабочую область. Он приобретает соответствующую форму: . Таким же образом изменяется форма курсора при выборе остальных шести кнопок строки инструментов.  Rotate out of plane (XY-вращение, свободное 3-х мерное)
Rotate out of plane (XY-вращение, свободное 3-х мерное)  Rotate in plane (Z-вращение, вращение в плоскости экрана)
Rotate in plane (Z-вращение, вращение в плоскости экрана)  Translate (перемещение в плоскости экрана)
Translate (перемещение в плоскости экрана)  Z-translate (перемещение в Z-направлении, вглубь экрана)
Z-translate (перемещение в Z-направлении, вглубь экрана)  Maghify/Shrink (масштабирование, увеличение / уменьшение)
Maghify/Shrink (масштабирование, увеличение / уменьшение)  Z-clipping planes (отрезание в z плоскости) При указании мышкой на кнопку появляется всплывающая подсказка, указывающая назначение выбранной кнопки. 2.3. Открытие файла В программе HyperChem Вы можете работать с молекулами, полученными тремя различными способами: 1. Инструментальные средства дают возможность создать двухмерную ( 2D ) структуру молекулы и затем преобразовывает ее с помощью моделепостроителя HyperChem в трехмерный ( 3D ) вид (см. далее п. 3). 2. Последовательный выбор из базы данных HyperChem уже готовых остатков аминокислот и нуклеотидов (нуклеозидов) позволяет создать белки и нуклеиновые кислоты. 3. Чтение данных с диска из файла в виде набора атомных и молекулярных координат, которые были сохранены в HyperChem в собственном формате файла (файл с расширением HIN ) или в фор-
Z-clipping planes (отрезание в z плоскости) При указании мышкой на кнопку появляется всплывающая подсказка, указывающая назначение выбранной кнопки. 2.3. Открытие файла В программе HyperChem Вы можете работать с молекулами, полученными тремя различными способами: 1. Инструментальные средства дают возможность создать двухмерную ( 2D ) структуру молекулы и затем преобразовывает ее с помощью моделепостроителя HyperChem в трехмерный ( 3D ) вид (см. далее п. 3). 2. Последовательный выбор из базы данных HyperChem уже готовых остатков аминокислот и нуклеотидов (нуклеозидов) позволяет создать белки и нуклеиновые кислоты. 3. Чтение данных с диска из файла в виде набора атомных и молекулярных координат, которые были сохранены в HyperChem в собственном формате файла (файл с расширением HIN ) или в фор-
мате белкового банка данных Brookhaven Protein Data Bank (файл ENT ), а также в ряде других форматов. Для открытия HIN файла с молекулярной структурой: 1. Левой кнопки мыши ( L-нажатие ) откройте меню File. 2. В меню выберите Open (Открыть). 3. Появляется список файлов в диалоговом окне, среди которых необходимо выбрать нужный и открыть его L-нажатием кнопки Открыть . Рис. 1.3. Диалог открытия файлов внутреннего формата HIN с примерами молекул из папки Samples Если файл, который Вы хотите увидеть, находится не в текущей директории Samples (встроенном в программу каталогесправочнике химический соединений), то просто измените директорию или диск. Чтобы появилась молекула фуллерена С 60 , как на рис. 1.4, выберите в директории Samples > Aromatic файл c60.hin . Вы можете также открывать файл двойным щелчком на имени файла. 2.4. Использование дисплейных установочных параметров HyperChem автоматически использует дисплейные установочные параметры с последнего сеанса работы. Выбрать или изменить дисплейные установочные параметры можно, используя команды пункта меню Display (Экран ).
5 quants / Лабораторный практикум Квантово-химическое моделирование соединений в пакете HyperChem Учебно-методическое пособие
Рис. 1.4. Пример открытого образца — молекулы фуллерена С 60 2.4.1. Использование подписей атомов и молекул Если, например, молекула отображается с подписями атомных зарядов или другими характеристиками, то удалить их можно следующим образом: 1. L-Нажатием кнопки мыши открыть в меню Display > Labels ( Этикетки ) (рис. 1.5). 2. В списке подписей атомов, связей и остатков автоматически помечено None ( Ничто ). 3. L-Нажатие на OK отменяет подписи атомов. Рис 2.5 . Выбор подписи атомов и пример молекулы нитробензола с отображением зарядов атомов Чтобы использовать другие подписи атомов (символы элементов, заряды, номера, массы и т.п.) необходимо в диалоговом окне пометить интересующий вас вариант. Диалог закрывается, и атомы мо- 11
лекулы помечаются выбранными химическими символами элементов, зарядами и пр. Используя пункт меню Display > Colors Atom … можно изменять цвета атомов со стандартных (по умолчанию) цветов на собственные варианты. 2.4.2. Использование различных молекулярных отображений Чтобы изменить изображения молекулярной системы: 1. Выбрать Renderings ( Отображение ) в меню Display . 2. В диалоговом окне выбрать вкладку Rendering method и пометить, например, Balls ( Шары ) (рис. 1.6). 3. Затем можно на вкладке Balls изменить размер радиуса шаров (рис. 1.6). Рис. 1.6. Выбор метода отображения – Balls (шары) Если нажать ОК, будет получено приближенное объемное изображение молекулы (рис. 1.7). Однако вы уже не увидите подписи отдельных атомов. Нажатие клавиши [ F2 ] позволяет восстановить параметры изображения молекулы, выбранные ранее (рис. 1.8). Это — эквивалент выбора Last Rendering (Последнее отображение ). Если в диалоговом окне Rendering method выбрать Balls & Cylinders (Шары и цилиндры ) и подобрать радиус шаров и цилиндров, вы получите изображение как на рис. 1.9. Такое представление, по мнению автора, лучше всего демонстрирует форму молекулы.
Рис. 1.7. Способы отображения молекул: Sticks («черточки») и Balls («шары»)
| Рис. 1.8 . Способы отображения мо- | Рис. 1.9. Способ отображения моле- |
| лекул: Sticks с обозначением символов | кул Balls & Cylinders |
| атомов |
С остальными способами отображения молекулярной структуры Вы познакомитесь самостоятельно. 2.5. Выход из HyperChem Выберите Exit в меню File . Программа может предупредить Вас относительно сохранения изменений в файле. Щелкните на кнопке « No », если Вы не желаете сохранить последнюю геометрию молекулы, или на кнопке « Yes » в обратном случае. При этом в файле Hin не сохраняются результаты расчета, такие как электронная структура и свойства, колебательный спектр и другие молекуляр- 13

ные свойства. Так что, в любом случае после открытия сохраненного файла придется выполнить расчет заново. 3. Техника построения и редактирования объектов После старта HyperChem желательно расширить окно программы на полный экран. В большой рабочей области гораздо удобнее строить молекулы. 3.1. Очистка рабочей области HyperChem Если у Вас в данный момент открыта предыдущая молекулярная структура, то очистить рабочее поле можно следующим образом. Выберите в меню File ( Файл ) пункт New ( Новый ). Появляется диалоговое окно (рис. 1.10), в котором содержится вопрос: «Хотите ли вы сохранить текущие изменения в данном файле?» Нужно выбрать вариант « Нет », если Вам не нужно сохранить текущие изменения. HyperChem очистит рабочую область. Рис. 1.10. Диалог сохранения изменений в файле 3.2. Рисование отдельных атомов До сих пор Вы учились отображать отдельные молекулы, читая их координаты из файла HIN . Покажем методы рисования и редактирования, которые используются для создания 2-D эскиза молекул. 1. Выберите в меню Build пункт Default element (Элемент по умолчанию) или выполните двойной щелчок на кнопке инструмен- та Draw (Рисовать). При этом запустится диалоговое окно Element Table – своего рода периодическая таблица элементов Менделеева (рис. 1.11). 2. Для выбора элемента курсор мыши наведите на желаемый элемент и щелкните левой клавишей.

Рис. 1.11. Диалог выбора элементов (Element Table) 3. Если нажать на кнопку Properties . , откроется окно, содержащее информацию о свойствах выбранного элемента (рис. 1.12). Рис. 1.12. Информация об элементе N (азот) 4. Для установления произвольной валентности атома в определенных связях нужно отметить Allow Arbitrary Valence (Допустить произвольную валентность). Опция Explicit hydrogens (Добавить водороды) служит для автоматического рисования атомов водорода. Мы ее не будем использовать. 5. После выбора первого элемента не закрывайте Element Table . 6. Переместите курсор в рабочую область. При этом он приобре- тает форму прицела . Если форма курсора другая, то выберите инструмент создания атомов Draw . 15
7. Разместите в поле построения выбранный атом, нажав однократно на левую кнопку мыши. Так можно разместить на любом расстоянии друг от друга неограниченное число атомов одного элемента, как, например, показано на рис. 1.13. 8. Для смены элемента необходимо в окне Default Element щелкнуть по нему левой кнопкой мыши и т.д. 3.3. Рисование связей 1. Для соединения двух атомов между собой «прицел» мыши наведите на центр атом и, не отпуская левой клавиши (L-протяжка), проведите линию до центра другого атома. После этого обозначение атомов в виде кружков исчезнет, а вместо них появиться линия, соединяющая атомы, т.е. связь. Таким образом, все размещенные в пространстве атомы можно соединить в одну молекулу посредством химических связей (рис. 1.14). Рис. 1.13 . Отдельные атомы углерода на рабочем поле (заготовка для создания бензольного кольцо и алифатической цепочки) 2. Существует альтернативный способ построения молекулы. После того, как Вы разместили один атом в поле построения, не отпуская левой клавиши (L-протяжка) проведите от него линию до места, где должен находиться второй такой же атом и, не отпуская кнопки мыши. Это будет позиция второго атома этого же элемента. 3. При построении молекул необходимо учитывать, что не все из них имеют плоскую структуру, поэтому для размещения отдельных атомов в пространстве под определенным углом относительно друг

друга молекулу можно разворачивать, выбрав мышью инструмент Rotate out of plane . 4. Чтобы химическую связь сделать кратной (двойной (рис. 1.14), тройной, полуторной), прицел необходимо разместить рядом с линией, изображающей одинарную связь и однократно (двукратно для ароматической) нажать на левую клавишу мыши. При этом в нижней строке появится соответствующая надпись ( single, double, triple, aromatic, см. таблицу 1.1). Рис. 1.14. Связанные атомы углерода 5. Для понижения порядка связи, удаления неверных связей или лишних атомов используют R-щелчок клавишей мыши по атомам или связям. При удалении связи будут удалены оба атома, участвующих в связи. Таблица 1.1. Примеры графического обозначения типов связей
| Ординарная | _____________________ |
| Двойная | =================== |
| Тройная | ≡≡≡≡≡≡≡≡≡≡≡≡≡≡≡≡≡≡≡ |
| Переменная (ароматическая) | ——————————— |
Если необходимо заменить один атом на другой (например, атом углерода на кислород), то после выбора в таблице нужного элемента, наведите курсор на место старого атома и щелкните по нему левой кнопкой мыши.

3.4. Работа с выделенными атомами (молекулами) Теперь перейдем к изучению методик выделения (выбора) отдельных объектов или всей вашей химической структуры. Удалить или копировать сразу несколько атомов или всю молекулу можно после их выделения. Для этой цели служит инструментальная кнопка Select (Выбор) (вложенные друг в друга пара кружочков). 1. Вначале необходимо выбрать параметры (режимы) выделения в меню Select . Можно выделять как отдельные Atoms ( Атомы) , Residues (О статки ), так и целые молекулы (Moleculs ), по отдель- ности или вместе. 2. После того, как Вы настроили параметры выделения (например, выбрали ٧ Atoms ), нужно навести курсор на выделяемый объект в рабочем поле и сделать L-щелчок кнопкой мыши. Выделенный атом помечается зеленным цветом (рис. 1.18). Обратите внимание: в окне диалога Preferences в меню File Вы также можете устанавливать цвет выделенных атомов, который Вы пожелаете.
| Рис. 1.18. Выделенный атом | Рис. 1.19. Выделенная связь между |
| атомами углерода 7 и 8 |
3. Таким же образом можно выделить связь между двумя атомами. В этом случае в параметрах выделения в меню Select должны быть обязательно отмечены Atoms и Multiple Selections . Для выделения связи просто щелкните подряд по двум соседним атомам или по центру связи (рис. 1.19). 18
Выделенные фрагменты можно удалить или запомнить в буфер с последующим извлечением из него (соответствующие кнопки на меню или разделы в меню Edit ). Режим Select Sphere в меню Select позволяет при выделении добавлять все последующие выделенные объекты к уже выделенным. Выделенные фрагменты можно также отдельно перемещать или вращать на рабочем поле, выбрав соответствующие инструменты и “захватив” их правой кнопкой мыши (только для этого необходимо в меню Fail > Preferences > Tool отменить ( ) whole molecular translation ). Быстро выделить всю молекулу можно однократным щелчком левой кнопкой по свободному пространству на рабочем поле или двойным щелчком по любому атому молекулы. Для отмены любого выделения поместите курсор в пустой области экрана и щелкните правой кнопкой мыши. Чтобы отменить выделение отдельного атома, фрагмента или связи, курсор в форме наводят на выделенный объект и однократно щелкают правой клавишей мыши. Важное замечание : Никогда не забывайте отменять выделения, если это специально не требуется для дальнейшей работы, т.к. все дальнейшие действия и операции, включая расчет, будут применяться только к выделенному фрагменту. 3.5. Выбор группы атомов При выборе произвольной группы атомов они должны быть заключены в нарисованный мышью воображаемый прямоугольник. 1. В меню Select не должно быть помечено Select Sphere . 2. Выберите точку в пустой части рабочей области, проведите от нее линию, одновременно удерживая левую и правую клавиши мыши (LR -протяжка) к выделяемым атомам. При этом будет рисоваться прямоугольник, отображающий границу области выбора. Продолжайте LR-перемещение, пока несколько атомов не будут находиться в прямоугольнике (как на рис. 1.20). 3. Отпустите кнопки мыши, и все атомы области выбора окажутся выделенными (рис. 1.21). 2. В этом же окне можно выделить вторую группу атомов. Для этого в меню Select необходимо отметить ( ) Multiple Selections . После чего таким же образом в прямоугольник заключить другую
группу атомов. При этом новые выделенные атомы добавятся к предыдущим.
| Рис. 1.20. Выбор группы атомов | Рис. 1.21. Выбранная группа атомов |

Напоминаем, чтобы выделить все атомы нужно в пустом пространстве рабочей области поместить курсор и однократно щелкнуть левой кнопкой мыши. Для отмены всех выделений нужно однократно нажать на правую кнопку мыши, когда курсор находится в пустом пространстве рабочей области. 3.6. Удаление атомов или связей Для удаления одного атома или связи: 1. Произведите однократный щелчок на инструментальном изо- бражении (Draw) на панели инструментов. 2. Наведите курсор на удаляемый объект и произведите R- щелчок. Атом или связь исчезнут. Для удаления нескольких атомов или связей: Посредством LR-протяжки заключите в пространство внутри прямоугольника атомы и связи, которые подлежат удалению В меню Edit ( Редактирования ) выберите Clear (Очистить ) . Появится диалоговый блок, где будет задан вопрос: » Вы действи- тельно хотите удалить выбор ?». Нужно нажать «Да». 3.7. Копирование объектов через буфер обмена 1. В меню Select отметьте Atoms . 2. Щелчком мыши в рабочем поле выделите атом или связь.
Как поменять фон в hyperchem
Программа HyperChem
Документация к программе HyperChem на английском языке находится в файлах CDK.pdf , GetStart.pdf , Referenc.pdf . Для читателей, абсолютно незнакомых с методами и программами квантовой химии и молекулярной динамики в данной Главе приводится некий “курс молодого бойца”, который позволяет легко начать работу с программой HyperChem.
После установки и запуска программы на мониторе появляется окно (вне зависимости от версии программы):
Части окна HyperChem
Строка названия показывает имя файла, с которым Вы работаете. Если Вы работаете во вновь созданном файле, имя появляется как untitled .
Строка меню содержит имена других HyperChem меню:
File Файл,
Edit Редактирование,
Build Построение объектов,
Select Выделение,
Display Отображение,
Databases Базы данных,
Setup Установки,
Compute Расчет,
Cance l Отмену,
Script Сценарий,
Help Подсказку.
Панель инструментов Левая сторона строки инструментов содержит восемь клавиш инструментальных средств, которые Вы используете, чтобы построить, выбрать, отобразить, и переместить атом или молекулу (см. ниже «Использование мыши»).
Строка состояния показывает текущую информацию, такую как количество атомов в молекуле, которое к настоящему времени отображается, состояние вычисления, энергию или величину градиента. Когда Вы выбираете пункт меню, краткое описание пункта появляется в строке состояния.
Кнопки управления меню содержат команды, позволяющие менять размеры, перемещать, расширять и закрывать окно HyperChem. Они также содержат команду Ключа, которая позволяет Вам активизировать другие окна.
Использование мыши
Работа в HyperChem по большей части происходит при помощи мыши. Она используется следующим образом:
1. Точка — перемещение точки (скольжение), при этом курсор указывает, что Вы хотите выбрать в окне HyperChem.
2. L-Нажатие — щелчок левой кнопкой мыши.
3. R-Нажатие — щелчок правой кнопкой мыши. Обычно, R-нажатие имеет противоположный эффект L-нажатию.
4. Двойное нажатие — быстрый двойной щелчок левой кнопкой мыши.
5. L-протяжка или R-протяжка — нажатие на левую или правую кнопку мыши, и перемещение, не отпуская кнопки, (скольжение) курсора в новую позицию в рабочей области.
6. LR-протяжка — нажатие и удержание левой кнопки мыши, затем правой кнопки и перемещение курсора в новую позицию в рабочей области.
7. RL-протяжка — так же, как LR-протяжка, но вначале нажатие правой кнопки мыши.
Курсор мыши способен изменять форму. Чтобы это увидеть:

1. Укажите на инструментальное изображение ( Draw ) нажмите левую клавишу.
2. Переместите курсор в рабочую область. Курсор приобретает соответствующую форму: Å .

3. Укажите на инструментальное изображение ( Select ) и нажмите левую клавишу.
4. Переместите курсор в рабочую область. Он приобретает соответствующую форму: ¥ . Таким же образом изменяется форма курсора при выборе остальных шести клавиш строки инструментов.

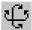




Клавиатурные альтернативы
HyperChem обеспечивает стандартные альтернативы клавиатуры
Чтобы открыть меню, использующее клавиатурную альтернативу, нажмите одновременно кнопку [Alt] и клавишу [S]. Открывается меню Выбора ( Select ).
Таким образом, можно открыть и все остальные меню, нажимая одновременно с [Alt] на клавишу первой буквы названия (F для меню File и т.д.).
Чтобы закрыть меню: нажмите [Alt] или клавишу [Esc].
Чтобы выбрать один из пунктов уже открытого меню: нажмите одновременно на кнопку [Alt] и клавишу, соответствующую первой букве названия пункта меню (A для пункта Atom).
Кратчайшие клавиатурные пути
HyperChem имеет различные кратчайшие клавиатурные пути.
[Ctrl] + [N] — создать новый файл ( New на Файловом меню).
[Ctrl] + [O] — открыть файл ( Open на Файловом меню).
[Ctrl] +[S] — сохранить ( Save )
[Ctrl] + [X] — вырезать ( Cut )
[Ctrl] + [C] — копировать ( Copy )
[Ctrl] + [V] — вставить ( Past )
[Alt] + [F4] — выход ( Exit )
[F4] — построение изоповерхностей
[F9] — копирование всего изображения ( Copy ../book/image(III)/Image ) в меню Редактирования.
[Esc] — отмена.
Открытие файла образца Sample File
В программе HyperChem Вы можете работать с молекулами тремя способами:
1. Инструментальные средства дают возможность создать двухмерную ( 2D ) структуру молекулы и затем преобразовывает ее с помощью моделепостроителя HyperChem в трехмерный ( 3D ) вид.
2. Выбор остатков позволяет последовательно выбрать из HyperChem/Lite уже готовые остатки аминокислот и нуклеотидов (нуклеозидов), чтобы создать белки и нуклеиновые кислоты.
3. Чтение: набор атомных и молекулярных координат, которые были сохранены в HyperChem формате входного файла (файл HIN ) или формате Белкового банка данных Brookhaven Protein Data Bank (файл ENT ), а также в ряде других форматов.
Открытие HIN файла
1. Переместить указатель мыши в верхнюю левую часть окна на меню File.
2. Нажатием левой кнопки мыши ( L-Нажатие ) откройте файловое меню
3. В диалоговом блоке меню выберите Open (Открыть )
4. Появляется список файлов, среди которых необходимо выбрать нужный и L-Нажатием кнопки мыши открыть его.

Использование дисплейных установочных параметров
HyperChem автоматически использует дисплейные установочные параметры с последнего сеанса работы. Выбрать дисплейные установочные параметры можно, используя пункт меню Display ( Отображение ).
Использование подписей атомов и молекул
Если молекула отображается с подписями атомных зарядов и пр., удалить их можно следующим образом:
1. L-Нажатием кнопки мыши открыть Labels ( Этикетки ).
2. В списке подписей атомов автоматически помечено None ( Ничто ).
3. L-Нажатие на OK отменяет подписи атомов. Диалоговый ящик закрывается, и подписи не отображаются.
Чтобы использовать подписи необходимо в диалоговом окне пометить интересующее и нажать ОК . Диалоговый ящик закрывается, и молекула помечается выбранными символами (химическими символами элементов, зарядами и пр.).
Использование молекулярных изображений
Чтобы изменить изображения молекулярной системы:
1. Выбрать Renderings в Display меню .
2. В диалоговом меню выбрать и пометить, например, Bolls ( Шары ).
3. В верхней части диалогового окна нажать на клавишу Bolls , которая открывает лист параметров

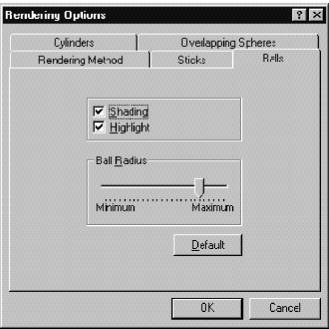
4. Если удалить метку ( Ö ) Shading ( Затенения ) и нажать ОК , будет получено приближенное, но быстро обрабатываемое объемное изображение молекулы.
5. Если же отметить Shading и затем нажать OK , это даст пространственно заполненное изображение.

6. Если в диалоговом меню выбрать Ö Sticks & Dots ( Стержни и точки ). Такое представление хорошо демонстрирует форму молекулы.
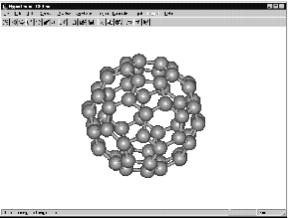
Нажатие клавиши [F2] позволяет восстановить параметры изображения молекулы, выбранные ранее. Это — эквивалент выбора Last Rendering ( Последний раз ) в дисплейном меню.
© И н с т и т у т Ф и з и к и
им. Л.В.Киренского 1998-2007
[an error occurred while processing this directive]